Research in Aging Cell indicates that blood levels of particular small non-coding RNAs, which regulate gene expression, may influence how long a person lives.
Investigators evaluated 828 small non-coding RNAs in blood samples from 1,271 community-dwelling older adults 71 years of age and older who were participating in an ongoing study. They then used machine learning to develop a model that could predict survival at 2, 5, and 10 years based on baseline small non-coding RNAs, age, and clinical variables (demographics, lifestyle, mood, physical function, standard clinical laboratory tests, lipid and metabolite levels, and medical conditions).
The test worked especially well for predicting survival over the next 2 years. “One surprising finding involved a group of small non-coding RNA molecules called piRNAs”, said co–corresponding author Virginia Byers Kraus, MD, PhD, of the Duke Molecular Physiology Institute. Scientists have long known that piRNAs help protect DNA in reproductive cells, but their role in the rest of the body is still a mystery. In this study, nine piRNAs, all reduced in longer-lived individuals, were identified as potential therapeutic targets to prolong longevity.
“These results suggest that simple blood tests measuring piRNAs might one day help doctors better understand health and aging – and possibly even guide new treatments to help people live longer, healthier lives,” said Dr Byers Kraus.
Rare diseases each affect relatively small numbers of people, but collectively they impact more than 300 million individuals worldwide across over 7000 known conditions, with 70% of these starting in childhood.1 For many patients and families, the reality is often long diagnostic journeys, uncertainty and ongoing challenges in accessing treatment and support.
This year’s global Rare Disease Day theme: “More Than You Can Imagine,” highlights the often unseen challenges faced by rare disease communities and the need for more equitable healthcare systems for people living with rare conditions worldwide.
Ahead of Rare Disease Day 2026, observed globally on 28 February, Sanofi South Africa is reaffirming its commitment to improving outcomes for people living with rare diseases through ongoing research, collaboration to improve access to treatment, and engagement in policy and advocacy discussions that support patients and caregivers.
According to Monique Nel, Medical Adviser for Rare Diseases at Sanofi South Africa, rare diseases demand a long-term mindset. “Patient populations may be small, but that makes every data point even more valuable. Building evidence takes time, yet each insight brings us closer to understanding these conditions and the unique needs of patients – enabling us to deliver better care.”
Research remains essential in rare diseases, where evidence is often limited and every patient experience matters. Global disease registries, such as the Global Gaucher Registry, allow clinicians and researchers to collect real-world data that deepens understanding of how conditions present across different regions and healthcare settings. Participation from South African patients helps ensure local experiences are reflected in global research.
“For me, equity starts with representation,” says Nel. “Patients are not the same everywhere. Genetics, environment, and healthcare systems all shape how a disease presents and progresses. If our research doesn’t reflect the diversity of the populations we serve, we risk missing a critical part of the picture.”
From scientific progress to real-world access
Innovation can transform outcomes for people living with rare diseases, but scientific progress only matters if patients can actually reach and stay on treatment.
Rare disease therapies are often complex and highly specialised, which means access depends on collaboration across clinicians, funders, policymakers and industry. The focus is increasingly on sustainable solutions that support affordability, continuity of care and long-term patient support.
“Access isn’t only about availability,” says Nel. “It’s also about what happens after treatment starts – whether patients can continue therapy, feel supported, and navigate their care with confidence.”
Strengthening policy and advocacy
Policy and legislative frameworks play an important role in shaping diagnosis, treatment pathways and long-term patient support. Ongoing engagement between stakeholders is essential to strengthen South Africa’s rare disease landscape and ensure decisions reflect real patient needs.
Patient voices are becoming increasingly important in policy and reimbursement discussions, offering insights that clinical data alone cannot provide.
“Patients and caregivers become experts through their own lived experiences,” says Nel. “Listening to their voices is what enables us to design better systems and ultimately deliver better care.”
“When we say rare diseases impact lives more than you can imagine, we’re talking about the invisible barriers patients face before they ever receive care,” says Nel. “Healthcare systems matter because they determine how quickly families find answers, how care is funded, and whether patients are truly included.”
She notes that South Africa’s constitutional commitment to healthcare, together with opportunities created through National Health Insurance, presents an important moment to strengthen support for rare disease communities.
South Africa’s support for the May 2025 rare diseases resolution at the World Health Assembly followed advocacy by Rare Diseases South Africa, which engaged the Department of Health and Health Minister Aaron Motsoaledi, calling for rare diseases to be recognised as a national health priority.
“Progress comes from sustained advocacy, partnership and action. Strong policy needs partners who understand that acting for patients means helping build systems that work for them,” says Nel.
Working with patient communities
As part of Rare Disease Day 2026, Sanofi South Africa is once again partnering with Rare Diseases South Africa (RDSA) to raise awareness around rare disease equity and amplify the lived experiences of patients and families. The collaboration focuses on education, awareness and encouraging meaningful dialogue around patient needs.
“Healthcare is a constitutional right in South Africa,” says Kelly du Plessis, CEO & Founder of Rare Diseases South Africa. “The opportunity now is to ensure rare disease patients are fully included in that promise. Equity means policies that don’t simply acknowledge rare diseases but actively prioritise them.”
RDSA remains an independent patient advocacy organisation, while the partnership supports awareness initiatives and responsible collaboration that strengthens patient-centred advocacy.
“We remain committed to working for patients, but we’ve learned to do that more effectively by collaborating with patient societies,” says Nel. “That partnership approach is essential. Equity means ensuring every patient is heard, every voice contributes, and every partnership has the opportunity to drive better care.”
The partnership with RDSA aims to:
Increase understanding of rare diseases and their impact
Support patient-centred advocacy and awareness
Encourage informed dialogue across healthcare stakeholders
Highlight the importance of equity in research, access and policy
Both organisations agree that meaningful progress in rare diseases depends on collective action across patients, healthcare professionals, policymakers and industry partners.
Reference: 1. World Health Organization (WHO). Rare diseases: a global health priority for equity and inclusion. Seventy-eighth World Health Assembly, Draft Resolution A78/51, Fifth report of Committee A, 24 May 2025. Available from: https://apps.who.int/gb/ebwha/pdf_files/WHA78/A78_51-en.pdf
Bicuspid aortic valve (BAV) is a common congenital heart defect where the aortic valve has two leaflets (cusps) instead of the usual three, resulting in abnormal blood flow and development of aortic valve diseases such as aortic stenosis and incompetence. In addition, the BAV is sometimes accompanied by development of an enlarged aorta – the main artery in the body. Both the bicuspid aortic valve and an enlarged aorta often require cardiac surgery, usually after the age of 50 years. Despite this, only a limited number of genes have been associated with the disease and the molecular mechanisms remain unexplained in most cases.
In a new study aimed to further understand the genetic architecture of BAV, an international group of researchers led by Boston University Chobanian & Avedisian School of Medicine and Laval University in Quebec City, Canada, along with the Bicuspid Aortic Valve Consortium, the Genetic Aortic Network (a division of The Marfan Foundation) and participating Institutions, believe the condition is strongly influenced by the cumulative effect of variation in many different genes(polygenic contribution).
“We found that variation in 36 genetic regions increases the risk of a bicuspid aortic valve. These findings support the notion that bicuspid aortic valve disease is an inherited disease caused by a combination of many common genetic variants, not merely a single mutation in a single gene,” explains co-corresponding author Simon C. Body, MD, MPH, professor of anesthesiology at Boston University Chobanian & Avedisian School of Medicine.
From a group of 65 677 US, Canadian and European participants, the researchers performed a genome-wide association study (GWAS) meta-analysis on 9631 individuals with BAV. After identifying general genetic regions through GWAS, they used RNA sequencing to study gene activity (expression levels) in specific, relevant tissues.
They observed 36 regions with genetic variants associated with a bicuspid aortic valve, four of which had been previously identified. They prioritised 55 genes in these regions based upon expression in human aortic valve tissues from individuals who had surgery, then tested the effect of changing four selected genes, upon heart development in an experimental model, demonstrating that all four altered genes had effects on development of the valve. The researchers also looked at the effect of these genes in a statistical model finding a three-fold increase in risk for a BAV in individuals in the top 10% and association with aortic aneurysmal disease, a bulge in the aortic wall that can rupture. Some of these 36 genetic regions are also involved in aortic stenosis and aortic aneurysm development, which could lead to better prediction of these complications in people with BAV and point to biological mechanisms responsible for these joint effects.
According to the researchers, while these findings support the notion that BAV is an inherited disease, the findings do not currently support genetic testing, either prenatally or later in life, for predicting a bicuspid aortic valve. “Echocardiography and other imaging modalities remain the gold standard for diagnosis. In addition, the identified heritability supports performing screening echocardiography on first-degree relatives of a person with an identified bicuspid valve,” adds Body.
These findings appear online in the journal Circulation.
Study finds genetic contribution to human lifespan is about 50% – more than double previous estimates
Photo by Matteo Vistocco on Unsplash
What determines how long we live – and to what extent is our lifespan shaped by our genes? Surprisingly, scientists believed for decades that the heritability of human lifespan was relatively low compared to other human traits, standing at just 20 to 25%; some recent large-scale studies even placed it below 10%. Now, a new study from the Weizmann Institute of Science, published in Science, presents an entirely different picture. According to the findings, genetics accounts for about 50% of variation in human lifespan – twice as much, or more, than previously thought.
The study was led by Ben Shenhar from the lab of Prof Uri Alon of Weizmann’s Molecular Cell Biology Department.
“For many years, lifespan was attributed mainly to non-genetic factors, fuelling scepticism about genetic determinants of longevity”
Using mathematical models and analyses of three large twin databases from Sweden and Denmark – including, for the first time in this context, a dataset of twins who were raised apart – the researchers showed that earlier heritability estimates were masked by high levels of extrinsic mortality, such as deaths caused by accidents, infections and environmental hazards. Filtering out such extrinsic factors was impossible in historic datasets because they provided no information about the cause of death. To compensate for this limitation, the researchers developed an innovative framework that included mathematical simulation of virtual twins to separate deaths due to biological ageing from those caused by extrinsic factors. The new results are consistent with the heritability of other complex human traits and with findings from animal models.
Science Numbers
Up to age 80, the risk of dying from dementia shows a heritability of about 70% – far higher than that of cancer or heart disease.
The results have far-reaching implications for ageing research and public health. “For many years, human lifespan was thought to be shaped almost entirely by non-genetic factors, which led to considerable scepticism about the role of genetics in ageing and about the feasibility of identifying genetic determinants of longevity,” says Shenhar. “By contrast, if heritability is high, as we have shown, this creates an incentive to search for gene variants that extend lifespan, in order to understand the biology of ageing and, potentially, to address it therapeutically.”
An analysis of genetic data over nearly one million individuals shows that certain stretches of DNA, made up of short sequences repeated over and over, become longer and more unstable as we age. The study found that common genetic variants can speed up or slow down this process by up to fourfold, and that certain expanded sequences are linked to serious diseases including kidney failure and liver disease.
Why it matters
More than 60 inherited disorders are caused by expanded DNA repeats: repetitive genetic sequences that grow longer over time. These include devastating conditions like Huntington’s disease, myotonic dystrophy, and certain forms of ALS. Most people carry DNA repeats that gradually expand throughout their lives, but this instability and what genetic factors control it hadn’t been fully analysed within large biobanks. This study demonstrates that DNA repeat expansion is far more widespread than previously recognised and identifies dozens of genes that regulate this process, opening new avenues for developing treatments that could slow disease progression.
What the study did
Researchers from UCLA, the Broad Institute, and Harvard Medical School analysed whole-genome sequencing data from 490 416 UK Biobank participants and 414 830 All of Us Research Program participants. They developed new computational methods to detect and measure DNA repeat lengths and instability from standard sequencing data. The team examined 356 131 polymorphic repeat locations across the genome, tracking how repeat lengths changed with age in blood cells and identifying genetic variants that influenced expansion rates. They also searched for links between repeat expansions and thousands of disease outcomes to discover previously unknown disease associations.
What they found
Common DNA repeats in blood cells expand as people age. The researchers identified 29 genetic locations where inherited variants modified DNA repeat expansion rates, with effects varying up to fourfold between individuals with the highest and lowest genetic risk scores. Interestingly, the same DNA repair genes had opposite effects on different repeats: variants that stabilised some repeats destabilised others. The study also discovered that expansions in the GLS gene, which have a prevalence of around 0.03%, were associated with 14-fold higher risk of severe kidney disease and 3-fold higher risk of liver diseases, representing a newly recognised repeat expansion disorder.
What’s next
The findings establish blood-based DNA repeat measurements as potential biomarkers for testing future therapies aimed at slowing repeat expansion in diseases like Huntington’s. The research team’s computational tools can now be applied to other large biobank datasets to discover additional unstable repeats and disease associations. Understanding why the same genetic modifiers have opposite effects on different repeats will require detailed mechanistic studies of how DNA repair processes vary across cell types and genetic contexts. The discovery of GLS repeat-associated kidney and liver disease suggests additional unrecognised repeat expansion disorders may be lurking in biobank data, waiting to be found.
From the experts
“We found that most human genomes contain repeat elements that expand as we age,” said Margaux L. A. Hujoel, PhD, lead author of the study and assistant professor in the Departments of Human Genetics and Computational Medicine at the David Geffen School of Medicine at UCLA. “The strong genetic control of this expansion, with some individuals’ repeats expanding four times faster than others, points to opportunities for therapeutic intervention. These naturally occurring genetic modifiers show us which molecular pathways could be targeted to slow repeat expansion in disease.”
A Scottish patient has become the first person in the world to receive a pioneering therapy aimed at improving outcomes for those having heart bypass surgery. The treatment involves precisely editing DNA in veins to be used during heart bypass surgery to boost the production of a protective protein.
The treatment could help extend the lifespan of blood vessels used during the surgery and significantly improve patient health, experts say.
Cause of failures in bypass surgery
Heart bypass surgery – an operation to improve blood flow to the heart – is a life-saving treatment for patients with coronary heart disease.
The process typically uses one artery and two or more veins as bypass grafts – healthy blood vessels used to bypass a narrowed or blocked artery – creating a new route for blood to flow.
Vein grafts used in this type of surgery can fail because they are not naturally designed to withstand the high pressure of blood flow from the heart.
Protecting vein grafts
The PROTECT study, led by NHS Greater Glasgow and Clyde and the University of Glasgow in collaboration with NHS Golden Jubilee and the University of Edinburgh, is trialling a new gene therapy designed to support newly grafted blood vessels.
The treatment will introduce a gene, which produces a protein called TIMP-3, into the vein to be grafted.
TIMP-3 is involved in tissue remodelling. Higher levels of the protein could help to prevent thickening and blockage of the blood vessel over time, scientists say.
Exciting milestone
The research team has developed a way to treat the graft directly at the time of surgery, safely and efficiently delivering the gene therapy to the affected tissue before grafting into the heart.
It is hoped the treatment will help to extend a patient’s healthy life expectancy and reduce the need for further surgeries, experts say.
In a groundbreaking study published in the Proceedings of the National Academy of Sciences (PNAS), scientists at Pacific Northwest Research Institute (PNRI) have overturned a long-held belief in genetics: that inheriting two harmful variants in the same gene always worsens disease. Instead, the team found that, in many cases, two harmful variants can actually restore normal protein function.
The research focused on a human enzyme called argininosuccinate lyase (ASL), which plays a critical role in removing toxic ammonia from the body. Variants in ASL that decrease its activity cause one of the urea cycle disorders, a set of rare and potentially life-threatening metabolic diseases.
By experimentally measuring the functional impact of several thousand individual variants and variant combinations, PNRI researchers discovered that over 60% of pairs that were individually damaging could, together, bring enzyme activity back to healthy levels.
“This work shows that genetic variants don’t act independently in many important cases,” said Michelle Tang, PhD, PNRI Staff Scientist and lead author of the study. “For a defined group of genes, the default assumptions we use to predict disease risk simply don’t hold.”
This phenomenon is known as intragenic complementation. It occurs when damage caused by one variant is offset by another variant in a different part of the same protein. The mechanism was first proposed in 1964 by Francis Crick and Leslie Orgel, but until now had not been tested systematically or shown to be common or predictable at scale.
To make these interactions predictable, the research team developed an AI-based model that accurately forecasted whether two variants would restore protein function. The model achieved nearly 100% accuracy in predicting intragenic complementation in ASL as well as in a second human enzyme, fumarase, suggesting these rules apply broadly across the human genome.
“We’ve shown that, in many cases, two damaging variants can work together to restore protein function,“ said Aimée Dudley, PhD, PNRI Senior Investigator who led the study. “This kind of genetic interaction is not an isolated exception, but a widespread and underappreciated way that variants can interact, especially in rare disease contexts.”
The researchers estimate that approximately 4% of human genes have the structural features that allow this type of interaction. For these genes, standard genetic predictions can overestimate disease risk, particularly for people who carry two different variants in the same gene.
Cedars-Sinai scientists have developed an experimental drug that repairs DNA and serves as a prototype for a new class of medications that fix tissue damage caused by heart attack, inflammatory disease or other conditions.
“By probing the mechanisms of stem cell therapy, we discovered a way to heal the body without using stem cells,” said Eduardo Marbán, MD, PhD, executive director of the Smidt Heart Institute at Cedars-Sinai and the study’s senior author. “TY1 is the first exomer – a new class of drugs that address tissue damage in unexpected ways.”
TY1 is a laboratory-made version of an RNA molecule that naturally exists in the body. The research team was able to show that TY1 enhances the action of a gene called TREX1, which helps immune cells clear damaged DNA. In so doing, TY1 repairs damaged tissue.
The development of TY1 has been more than two decades in the making. It started when Marbán’s previous laboratory at Johns Hopkins University developed a technique to isolate progenitor cells from the human heart. Like stem cells, progenitor cells can turn into new healthy tissue, but in a more focused manner than stem cells. Heart progenitor cells promote the regeneration of the heart, for example.
Later, at Marbán’s lab at Cedars-Sinai, Ahmed Ibrahim, PhD, MPH, discovered that these heart progenitor cells send out tiny molecule-filled sacs called exosomes. These sacs are loaded with RNA molecules that help repair and regenerate injured tissue.
Ahmed Ibrahim, PhD, MPH“Exosomes are like envelopes with important information,” said Ibrahim, who is associate professor in the Department of Cardiology in the Smidt Heart Institute and first author of the paper. “We wanted to take apart these coded messages and figure out which molecules were, themselves, therapeutic.”
Scientists genetically sequenced the RNA material inside the exosomes. They found that one RNA molecule was more abundant than the others, hinting it might be involved in tissue healing. The investigators found the natural RNA molecule to be effective in promoting healing after heart attacks in laboratory animals. TY1 is the synthetic, engineered version of that RNA molecule, designed to mimic the structure of approved RNA drugs already in the clinic. TY1 works by increasing the production of immune cells that reverse DNA damage, a process that minimises the formation of scar tissue after a heart attack.
“By enhancing DNA repair, we can heal tissue damage that occurs during a heart attack,” Ibrahim said. “We are particularly excited because TY1 also works in other conditions, including autoimmune diseases that cause the body to mistakenly attack healthy tissue. This is an entirely new mechanism for tissue healing, opening up new options for a variety of disorders.”
The investigators next plan to study TY1 in clinical trials.
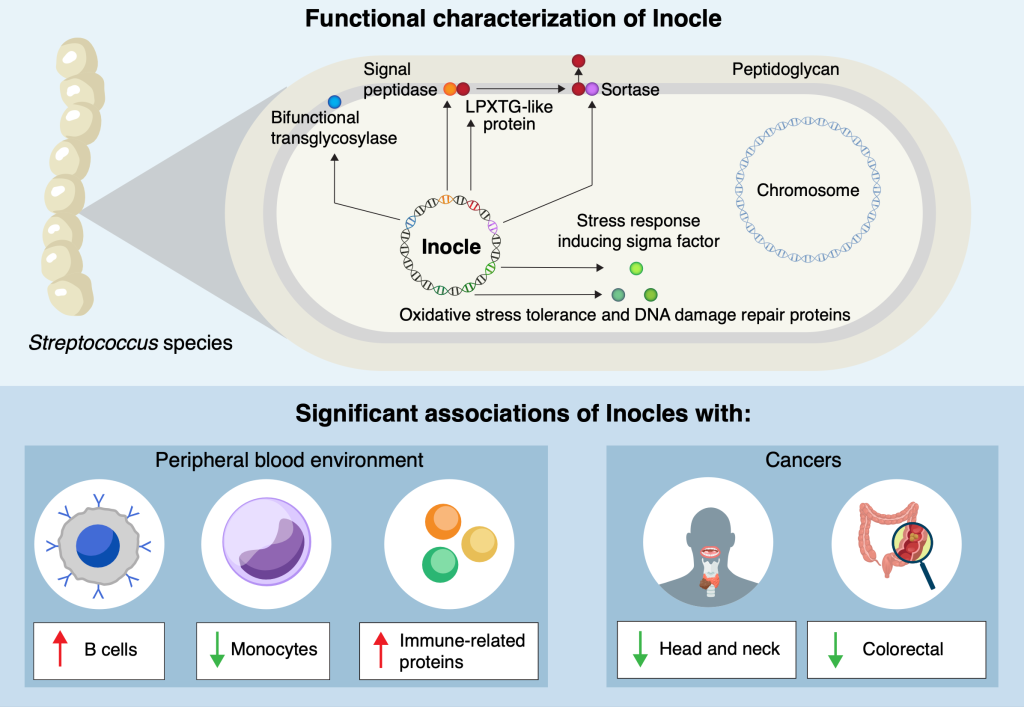
Researchers at the University of Tokyo and other institutions have made a surprising discovery hiding in people’s mouths: Inocles, giant DNA elements that had previously escaped detection. These appear to play a central role in helping bacteria adapt to the constantly changing environment of the mouth. The findings provide fresh insight into how oral bacteria colonise and persist in humans, with potential implications for health, disease and microbiome research.
You might think that modern medical science knows everything there is to know about the human body. But even within the last decade, small, previously unknown organs have been discovered, and there’s one area of human biology that is currently going through a research renaissance, the microbiome. This includes familiar areas such as the gut microbiome, but also the oral microbiome. Inspired in part by recent discoveries of extraneous DNA in the microbiome of soil, Project Research Associate Yuya Kiguchi and his team turned their sights to a large set of saliva samples collected by the Yutaka Suzuki Lab of the Graduate School of Frontier Sciences at the University of Tokyo. They wondered if they might find something similar in human saliva.
“We know there are a lot of different kinds of bacteria in the oral microbiome, but many of their functions and means of carrying out those functions are still unknown,” said Kiguchi. “By exploring this, we discovered Inocles, an example of extrachromosomal DNA – chunks of DNA that exist in cells, in this case bacteria, but outside their main DNA. It’s like finding a book with extra footnotes stapled to it, and we’re just starting to read them to find out what they do.”
Detecting Inocles was not easy, as conventional sequencing methods fragment genetic data, making it impossible to reconstruct large elements. To overcome this, the team applied advanced long-read sequencing techniques, which can capture much longer stretches of DNA. A key breakthrough came from co-first author Nagisa Hamamoto, who developed a method called preNuc to selectively remove human DNA from saliva samples, greatly improving the quality of sequencing long sections of other DNA. This allowed the researchers to assemble for the first time complete Inocle genomes, which turned out were hosted by the bacteria Streptococcus salivarius, though identifying the host itself was a difficult matter.
“The average genome size of Inocle is 350 kilobase pairs, a measure of length for genetic sequences, so it is one of the largest extrachromosomal genetic elements in the human microbiome. Plasmids, other forms of extrachromosomal DNA, are at most a few tens of kilobase pairs,” said Kiguchi. “This long length endows Inocles with genes for various functions, including resistance to oxidative stress, DNA damage repair and cell wall-related genes, possibly involved in adapting to extracellular stress response.”
The team aims to develop stable methods for culturing Inocle-containing bacteria. This will allow them to investigate how Inocles function, whether they can spread between individuals, and how they might influence oral health conditions such as cavities and gum disease. Since many Inocle genes remain uncharacterised, researchers will use a mixture of laboratory experiments and also computational simulations such as AlphaFold to predict and model the roles Inocles may play.
“What’s remarkable is that, given the range of the human population the saliva samples represent, we think 74% of all human beings may possess Inocles. And even though the oral microbiome has long been studied, Inocles remained hidden all this time because of technological limitations,” said Kiguchi. “Now that we know they exist, we can begin to explore how they shape the relationship between humans, their resident microbes and our oral health. And there’s even some hints that Inocles might serve as markers for serious diseases like cancer.”
University of Hawaiʻi at Mānoa scientists have uncovered a direct link between a missing Y chromosome gene and male infertility. Their new research reveals that deleting this single gene in mice not only caused infertility but also disrupted hundreds of other genes vital for healthy sperm. The findings, published August 27 in Cell Death and Differentiation, offer significant implications for understanding reproductive health.
Using CRISPR gene-editing, the team created mice missing one or both versions. Males without both, known as Zfy double knockouts, were completely infertile, with severely abnormal or absent sperm.
“This work really pushes forward our understanding of how this important Zfy gene works,” said Ward. “We identified pathways and other genes that are affected and we can now study how exactly Zfy regulates them.”
To continue investigations, the researchers turned to assisted reproduction techniques pioneered at UH, including intracytoplasmic sperm injection (ICSI) and round spermatid injection (ROSI). This allowed them to examine the molecular consequences of Zfy loss.
When one gene disrupts hundreds
The results revealed that without Zfy, hundreds of genes became misregulated – some too active, others too weak. Many of these genes are responsible for sperm production, DNA packaging, and cell survival.
As a result, sperm precursor cells in the testes died off early, and the sperm that did form carried fragile DNA that wasn’t properly condensed.
The study details can be found in an article published in Cell Death and Differentiation, a leading peer-reviewed journal.