Hydrogen Sulphide Could be the Answer to Treating Nail Infections
Scientists at Bath and King’s College London have discovered that a common chemical could be used to develop a new treatment for difficult-to-treat nail infections.
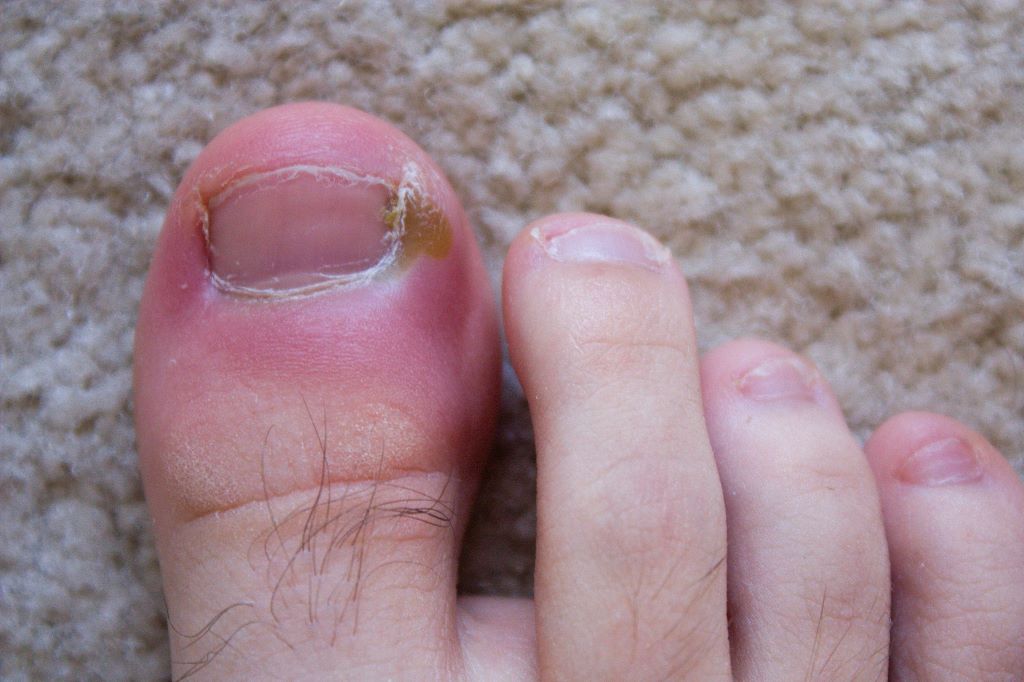
Hydrogen sulphide, the volcanic gas that smells of rotten eggs, could be used in a new treatment for tricky nail infections that acts faster but with fewer side effects, according to scientists at the University of Bath and King’s College London (KCL).
Nail infections are mostly caused by fungi and occasionally by bacteria. They are very common, affecting between 4-10% of the global population, rising to nearly half those aged 70 or over.
These infections can lead to complications, particularly in vulnerable groups such as diabetics and the elderly, but are notoriously difficult to treat.
Current treatments include oral antifungals taken in pill form, and topical treatments which are applied directly to the nail.
Oral antifungals take around 2-4 months to act and are reasonably effective, but they carry risks of side effects, especially in patients with other medical conditions.
Treatments applied directly to the nail are safer, but they often take much longer to work, sometimes taking even years to work, and they frequently relapse or fail.
This is largely because it’s very difficult to get the drug to penetrate through the nail to where the infection resides.
Even the most effective topical treatments have relatively low cure rates, so there is a clear need for new therapeutic approaches that are safe, effective, and capable of reaching microbes embedded deep within the nail.
A team from the University of Bath and King’s College London has now found that hydrogen sulphide (H₂S), a small, naturally occurring gas, could be developed into a promising new treatment.
Previous work has shown that it penetrates the nail plate far more efficiently than existing topical drugs, and now the team has demonstrated that it has strong antimicrobial activity against a wide range of nail pathogens, including fungi that are resistant to common antifungal treatments.
In laboratory tests, the team used a chemical that breaks down to release the H₂S gas and found that it acts in a unique way, disrupting microbial energy production and triggering irreversible damage, ultimately killing the fungi.
The research is published in Scientific Reports.
Dr Albert Bolhuis, from the University of Bath’s Department of Life Sciences, said: “Thanks to its ability to efficiently reach the site of infection and its novel mode of action, we believe that a topically applied medicine containing hydrogen sulphide could become a highly effective new treatment for nail infections, which avoids the limitations of current therapies.
“Our research lays the foundation for a compelling alternative to existing treatments, with the potential to improve outcomes for patients suffering from persistent and drug-resistant fungal nail infections.”
Hydrogen sulphide is known for its pungent smell of rotten eggs, and has some toxicity, however researchers believe the amounts required are well below toxicity levels and the correct formulation will limit any unpleasant odours.
The research has so far only been done in vitro, but the team hopes to develop a treatment that could be used in patients in the next five years.
Professor Stuart Jones, Director of the Centre for Pharmaceutical Medicine Research at KCL said: “We are looking forward to translating these findings into an innovative topical product that can treat nail infection.”
Source: University of Bath