Gene Therapy Shows Promise for an Inherited Form of Cardiomyopathy
A new gene therapy appears to be safe in patients diagnosed with Friedreich ataxia cardiomyopathy, a progressive and fatal inherited cardiac disease, according to a phase 1 clinical trial led by Weill Cornell Medicine researchers. The treatment may also reduce heart damage, although further investigation is needed.
The results, published June 17 in JAMA Cardiology, indicated that an intravenous infusion of a healthy frataxin (FXN) gene was generally well tolerated and shows early signs of efficacy. These include a decrease in heart wall thickness – enlarged walls are a sign of cardiomyopathy – and reduced levels of troponin I, a marker of heart damage.
“This is a fatal disease, but this is a potential therapy, and our goal is FDA-approval,” said Dr Ronald G. Crystal, the study’s lead author, professor and chair of the Department of Genetic Medicine at Weill Cornell Medicine and a pulmonologist at NewYork-Presbyterian/Weill Cornell Medical Center.
What is Friedreich Ataxia?
Friedreich ataxia is caused by variants in the FXN gene, leading to decreased levels of the FXN protein, which is essential for energy production in cells. “The two most energy consuming organs in the body are your brain and the heart, so the disease is primarily a brain and heart disease,” Dr Crystal said.
It is an autosomal recessive hereditary disorder, meaning a person must inherit a faulty copy of the FXN gene from both parents. As many as one in 50 000 people in the United States are diagnosed with the disease, according to some reports.
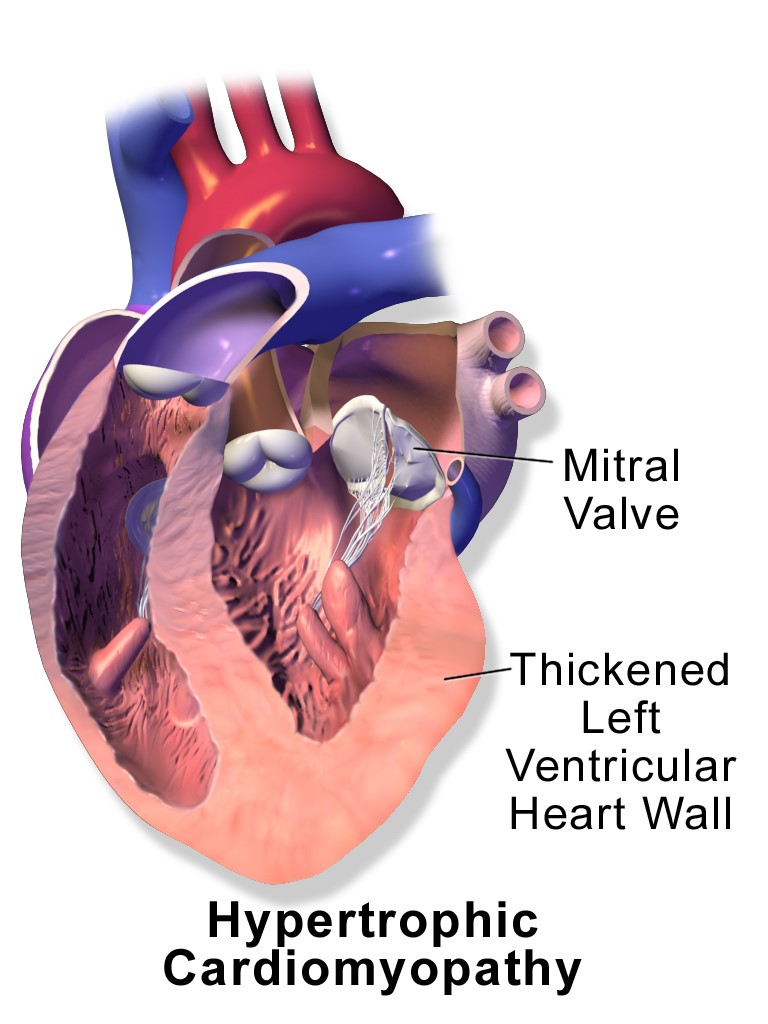
Nervous system symptoms typically begin in childhood and include problems with balance, walking and speaking. While neurologic disease is devastating for maintaining quality of life, most people with Friedreich ataxia develop heart disease, which is the cause of death in up to 65 percent of patients, according to reported estimates. Decreased FXN protein levels in the heart mean the heart cells don’t have the energy to beat normally. The muscle cells grow and the heart walls thicken, a condition known as hypertrophic cardiomyopathy, which can cause dangerous irregular heartbeats and heart failure.
The US Food and Drug Administration has approved only one other drug, omaveloxolone, to treat Friedreich ataxia. It slows the neurological symptom progression but does not address the direct genetic cause of the disease.
A New Gene Therapy
Based on promising preclinical research, Dr Crystal and his colleagues studied the safety and efficacy of the FXN gene therapy in 17 patients with Friedreich ataxia cardiomyopathy.
“We put the healthy FXN gene in a virus, called adeno-associated virus, which is given intravenously and likes to travel to the heart,” he said.
The researchers pooled data from two independent studies: nine patients were from a Weill Cornell Medicine study, funded by National Heart Lung Blood Institute, and eight were treated in a study by Lexeo Therapeutics, a clinical stage genetic medicine company founded by Dr Crystal. Weill Cornell Medicine Enterprise Innovation, which aims to accelerate the translation of scientific discoveries into patient impact, played a crucial role in launching Lexeo in 2020 and later licensed to it additional technology to further support the clinical trial.
In both studies, the patients received a one-hour infusion of the gene therapy and were evaluated from six to 36 months. Three different doses were tested among three groups of patients.
Overall, the drug was safe, causing four serious adverse events, which were all resolved. Three of these were possibly related to prednisone, an immunosuppression drug that patients took so their bodies did not attack the gene therapy.
In the Lexeo study, researchers took biopsies of the heart before therapy and three months after therapy and found that frataxin protein levels increased in cardiac tissue in all eight patients. Researchers also found that the left ventricular mass index, which is an MRI measurement of heart wall thickness, decreased, demonstrating that the treatment was therapeutic for cardiomyopathy.
Levels of troponin I, a structural protein of the heart that is released into the circulation when the heart is damaged, also decreased. Troponin I levels are typically high in patients with Friedreich ataxia cardiomyopathy.
Using the modified Friedreich Ataxia Rating Scale (mFARS), which assesses balance, coordination, speech, and limb function in patients, the researchers found that some neurological components of the disease stabilised. “But we’re unsure whether this was related to the gene therapy reaching the skeletal muscle or the brain,” Dr Crystal said. “That remains to be seen.”
Because most of the patients evaluated in this study had early cardiomyopathy, the researchers also hope to study the gene therapy in people who have a wider range of heart disease severity.
Source: Weill Cornell Medicine