Couple or siblings? New study may explain why we prefer partners who are similar to us. Photo by Daniil Onischenko on Unsplash
It is no secret that people are often drawn to romantic partners who seem similar to themselves. This tendency, called assortative mating, has been established in humans (Horwitz et al., 2023; Luo, 2017) as well as other species. Fish, for example, demonstrate the behaviour frequently (Jiang et al., 2013).
Assortative mating has also recently been in focus on social media with the viral Siblings or Dating game, where people guess whether two individuals who look alike are related or a couple.
The idea is well-founded in academic research. Humans have been observed to select partners with similar physical, personality, and demographic traits (Horwitz et al., 2023), which can impact the genetics of populations – creating subgroups that emphasise the presence of shared traits (Abdellaoui et al., 2015).
But selecting a partner like ourselves may not be solely determined by personal choice. A new study soon to be published in Psychological Science suggests that assortative mating can be explained relatively simply by looking at the inheritance of preferred traits and corresponding preferences for those traits.
Coauthors Kaitlyn Harper and Brendan Zietsch from the University of Queensland describe this scenario simply: If you are tall, you may have inherited tallness from one parent (say, your mother) and the preference for tallness in a romantic partner from your other parent (in this case, your father). The combination of those inherited traits means that you exist in the world as a tall person and are attracted to tall people.
The idea that preference for a particular trait could lead to genetic correlations has been discussed in previous research but is a newer concept for evolutionary psychology, especially in the context of assortative mating.
“The pieces were there, but they hadn’t been connected in this way before,” Harper said. “Agent-based modelling helped us connect the dots – by simulating populations, we could see that assortative mating naturally emerged without the need for additional assumptions or processes.”
She added that this research wouldn’t have been possible without an interdisciplinary mindset.
“The mechanism itself is familiar in evolutionary biology, but it wasn’t thought of as an explanation for assortative mating,” she said. “Making that connection only became possible when we looked across the two disciplines.”
To test this theory, the authors ran an agent-based model where partners are chosen according to heritable traits and preferences over 100 generations. They included models with and without selection pressure on the number of offspring within each generation to assess how the theory stands up under more naturalistic conditions.
They found that even with up to 10 preferences for traits in a partner, clear genetic correlations formed between traits and preferences for those traits, which resulted in the agents choosing partners similar to themselves. Models with selection pressure generated less-stable correlations, which the authors attribute to reduced variance in traits.
“The power of this finding is in its parsimony – it shows that a phenomenon which has puzzled researchers for decades can be understood through an explanation that was hiding in plain sight,” Harper said. “And because the mechanism is so general, it can also apply to assortative mating in animals, where many of the explanations proposed for humans wouldn’t make sense.”
Animals that hibernate are incredibly resilient. They can spend months without food or water, muscles refusing to atrophy, body temperature dropping to near freezing as their metabolism and brain activity slow to a crawl. When they emerge from hibernation, they recover from dangerous health changes similar to those seen in type 2 diabetes, Alzheimer’s disease, and stroke.
New genetic research suggests that hibernating animals’ superpowers could lie hidden in human DNA – with clues on how to unlock them, perhaps one day leading to treatments that could reverse neurodegeneration and diabetes.
A gene cluster called the “fat mass and obesity (FTO) locus” plays an important role in hibernators’ abilities, the researchers found. Intriguingly, humans have these genes too. “What’s striking about this region is that it is the strongest genetic risk factor for human obesity,” says Chris Gregg, PhD, professor in neurobiology and human genetics at University of Utah Health and senior author on the studies. But hibernators seem able to use genes in the FTO locus in new ways to their advantage.
The team identified hibernator-specific DNA regions that are near the FTO locus and that regulate the activity of neighbouring genes, tuning them up or down. The researchers speculate that adjusting the activity of neighbouring genes, including those in or near the FTO locus, allows hibernators to pack on the pounds before settling in for the winter, then slowly use their fat reserves for energy throughout hibernation.
Indeed, the hibernator-specific regulatory regions outside of the FTO locus seem crucial for tweaking metabolism. When the researchers mutated those hibernator-specific regions in mice, they saw changes in the mice’s weight and metabolism. Some mutations sped up or slowed down weight gain under specific dietary conditions; others affected the ability to recover body temperature after a hibernation-like state or tuned overall metabolic rate up or down.
Intriguingly, the hibernator-specific DNA regions the researchers identified weren’t genes themselves. Instead, the regions were DNA sequences that contact nearby genes and turn their expression up or down, like an orchestra conductor fine-tuning the volume of many musicians. This means that mutating a single hibernator-specific region has wide-ranging effects extending far beyond the FTO locus, explains Susan Steinwand, research scientist in neurobiology at U of U Health and first author on one of the studies. “When you knock out one of these elements – this one tiny, seemingly insignificant DNA region – the activity of hundreds of genes changes,” she says. “It’s pretty amazing.”
Understanding hibernators’ metabolic flexibility could lead to better treatments for human metabolic disorders like type 2 diabetes, the researchers say. “If we could regulate our genes a bit more like hibernators, maybe we could overcome type 2 diabetes the same way that a hibernator returns from hibernation back to a normal metabolic state,” says Elliott Ferris, MS, bioinformatician at U of U Health and first author on the other study.
Uncovering the regulation of hibernation
Finding the genetic regions that may enable hibernation is a problem akin to excavating needles from a massive DNA haystack. To narrow down the regions involved, the researchers used multiple independent whole-genome technologies to ask which regions might be relevant for hibernation. Then, they started looking for overlap between the results from each technique.
First, they looked for sequences of DNA that most mammals share but that had recently changed in hibernators. “If a region doesn’t change much from species to species for over 100 million years but then changes rapidly and dramatically in two hibernating mammals, then we think it points us to something that is important for hibernation, specifically,” Ferris says.
To understand the biological processes that underlie hibernation, the researchers tested for and identified genes that turn up or down during fasting in mice, which triggers metabolic changes similar to hibernation. Next, they found the genes that act as central coordinators, or “hubs,” of these fasting-induced changes to gene activity.
Many of the DNA regions that had recently changed in hibernators also appeared to interact with these central coordinating hub genes. Because of this, the researchers expect that the evolution of hibernation requires specific changes to the controls of the hub genes. These controls comprise a shortlist of DNA elements that are avenues for future investigation.
Awakening human potential
Most of the hibernator-associated changes in the genome appeared to “break” the function of specific pieces of DNA, rather than confer a new function. This hints that hibernators may have lost constraints that would otherwise prevent extreme flexibility in the ability to control metabolism. In other words, it’s possible that the human “thermostat” is locked to a narrow range of continuous energy consumption. For hibernators, that lock may be gone.
Hibernators can reverse neurodegeneration, avoid muscle atrophy, stay healthy despite massive weight fluctuations, and show improved aging and longevity. The researchers think their findings show that humans may already have the needed genetic code to have similar hibernator-like superpowers—if we can bypass some of our metabolic switches.
“Humans already have the genetic framework,” Steinwand says. “We just need to identify the control switches for these hibernator traits.” By learning how, researchers could help confer similar resilience to humans.
Genetic analysis of the early pandemic virus shows key adaptations to humans.
Creative artwork featuring colourised 3D prints of influenza virus (surface glycoprotein hemagglutinin is blue and neuraminidase is orange; the viral membrane is a darker orange). Note: Not to scale. Credit: NIAID
Researchers from the universities of Basel and Zurich have used a historical specimen from UZH’s Medical Collection to decode the genome of the virus responsible for the 1918-1920 influenza pandemic in Switzerland. The genetic material of the virus reveals that it had already developed key adaptations to humans at the outset of what became the deadliest influenza pandemic in history.
New viral epidemics pose a major challenge to public health and society. Understanding how viruses evolve and learning from past pandemics are crucial for developing targeted countermeasures. The so-called Spanish flu of 1918-1920 was one of the most devastating pandemics in history, claiming some 20 to 100 million lives worldwide. And yet, until now, little has been known about how that influenza virus mutated and adapted over the course of the pandemic.
More than 100-year-old flu virus sequenced
An international research team led by Verena Schünemann, a paleogeneticist and professor of archaeological science at the University of Basel (formerly at the University of Zurich) has now reconstructed the first Swiss genome of the influenza virus responsible for the pandemic of 1918-1920. For their study, the researchers used a more than 100-year-old virus taken from a formalin-fixed wet specimen sample in the Medical Collection of the Institute of Evolutionary Medicine at UZH. The virus came from an 18-year-old patient from Zurich who had died during the first wave of the pandemic in Switzerland and underwent autopsy in July 1918.
Three key adaptations in Swiss virus genome
“This is the first time we’ve had access to an influenza genome from the 1918-1920 pandemic in Switzerland. It opens up new insights into the dynamics of how the virus adapted in Europe at the start of the pandemic,” says last author Verena Schünemann. By comparing the Swiss genome with the few influenza virus genomes previously published from Germany and North America, the researchers were able to show that the Swiss strain already carried three key adaptations to humans that would persist in the virus population until the end of the pandemic.
Two of these mutations made the virus more resistant to an antiviral component in the human immune system – an important barrier against the transmissions of avian-like flu viruses from animals to humans. The third mutation concerned a protein in the virus’s membrane that improved its ability to bind to receptors in human cells, making the virus more resilient and more infectious.
New genome-sequencing method
Unlike adenoviruses, which cause common colds and are made up of stable DNA, influenza viruses carry their genetic information in the form of RNA, which degrades much faster. “Ancient RNA is only preserved over long periods under very specific conditions. That’s why we developed a new method to improve our ability to recover ancient RNA fragments from such specimens,” says Christian Urban, the study’s first author from UZH. This new method can now be used to reconstruct further genomes of ancient RNA viruses and enables researchers to verify the authenticity of the recovered RNA fragments.
Invaluable archives
For their study, the researchers worked hand in hand with UZH’s Medical Collection and the Berlin Museum of Medical History of the Charité University Hospital. “Medical collections are an invaluable archive for reconstructing ancient RNA virus genomes. However, the potential of these specimens remains underused,” says Frank Rühli, co-author of the study and head of the Institute of Evolutionary Medicine at UZH.
The researchers believe the results of their study will prove particularly important when it comes to tackling future pandemics. “A better understanding of the dynamics of how viruses adapt to humans during a pandemic over a long period of time enables us to develop models for future pandemics,” Verena Schünemann says. “Thanks to our interdisciplinary approach that combines historico-epidemiological and genetic transmission patterns, we can establish an evidence-based foundation for calculations,” adds Kaspar Staub, co-author from UZH. This will require further reconstructions of virus genomes as well as in-depth analyses that include longer intervals.
While the incidence of breast cancer is highest for white women, Black women are more likely to have early-onset or more aggressive subtypes of breast cancer, such as triple-negative breast cancer. Among women under 50, the disparity is even greater: young Black women have double the mortality rate of young white women.
Now, research from the University of Notre Dame is shedding light on biological factors that may play a role in this disparity. The study published in iScience found that a population of cells in breast tissues, dubbed PZP cells, send cues that prompt behavioural changes that could promote breast cancer growth.
Funded by the National Cancer Institute at the National Institutes of Health, the study set out to explore what biological differences in breast tissue could be related to early onset or aggressive breast cancers. Most breast cancers are carcinomas, or a type of cancer that develops from epithelial cells. In healthy tissue, epithelial cells form linings in the body and typically have strong adhesive properties and do not move.
The researchers focused on PZP cells as previous studies had shown that these cells are naturally and significantly higher in healthy breast tissues of women of African ancestry than in healthy breast tissues of women of European ancestry. While PZP cell levels are known to be elevated in breast cancer patients in general, their higher numbers in healthy, African ancestry tissues could hold clues to why early-onset or aggressive breast cancers are more likely to occur in Black women.
“The disparity in breast cancer mortality rates, particularly among women of African descent, is multifaceted. While socioeconomic factors and delayed diagnosis may be contributing factors, substantial emerging evidence suggests that biological and genetic differences between racial groups can also play a role,” said Crislyn D’Souza-Schorey, the Morris Pollard Professor of Biological Sciences at Notre Dame and corresponding author of the study.
The study showed how PZP cells produce factors that activate epithelial cells to become invasive, where they detach from their primary site and invade the surrounding tissue.
For example, a particular biological signaling protein known as AKT is often overactive in breast cancers. This study showed that PZP cells can activate the AKT protein in breast epithelial cells, which in part allows them to invade the surrounding environment. PZP cells also secrete and deposit certain proteins outside the cell that guide the movement of breast epithelial cells as they invade.
Overall, the results of the study emphasize multiple mechanisms by which PZP cells may influence the early stages of breast cancer progression and their potential contribution to disease burden.
The researchers also looked at how a targeted breast cancer drug, capivasertib, which inhibits the AKT protein, impacted PZP cells and found it markedly reduced the effects of the PZP cells on breast epithelial cells.
“It’s important to understand the biological and genetic differences within normal tissue as well as tumours among racial groups, as these variations could potentially influence treatment options and survival rates. And consequently, in planning biomarker studies, cancer screenings or clinical trials, inclusivity is important,” said D’Souza-Schorey, also an affiliate of Notre Dame’s Berthiaume Institute for Precision Health and Harper Cancer Research Institute.
Genetic ancestry is much more complicated than how people report their race and ethnicity. New research, using data from the National Institutes of Health’s (NIH) All of Us Research Program, finds that people who identify as being from the same race or ethnic group can have a wide range of genetic differences. The findings are reported in the American Journal of Human Genetics, a Cell Press journal.
As doctors and researchers learn more about how genetic variants influence the incidence and course of human diseases, the study of genetic ancestry has become increasingly important. This research is driving the field of precision medicine, which aims to develop individualised healthcare.
People whose ancestors came from the same part of the world are likely to have inherited the same genetic variants, but self-identified race and ethnicity don’t tell the whole story about a person’s ancestors. NIH’s All of Us Research Program was created in part to address this puzzle and to learn more about how genetic ancestry influences human health.
In the current study, the investigators looked at the DNA of more than 230 000 people who have volunteered to share their health information for All of Us. They compared it to other large DNA projects from around the world using a technique called principal component analysis (PCA) to visualize population structure and help identify genetic similarity between individuals and groups of people. This analysis showed that people in the US have very mixed ancestry, and their DNA doesn’t always match the race or ethnicity they write on forms. Instead of falling into clear groups based on race or ethnicity, people’s genetic backgrounds show gradients of variation across different US regions and states.
This is especially significant for people who identify as being of Hispanic or Latino origin. These people have a wide-ranging blend of ancestries from European, Native American, and African groups. Importantly, genetic ancestry among these people varies across the US in part because of historic migration patterns. For example, Hispanics/Latinos in the Northeast are more likely to have Caribbean (and thus African) ancestry, and those in the Southwest are more likely to have Mexican and Central American (and thus Native American) ancestry.
One specific discovery was that ancestry was significantly associated with body mass index (BMI) and height, even after adjusting for socio-economic differences. For example, West and Central African ancestries were associated with higher BMI, whereas East Africa ancestry was associated with lower BMI. There were similar findings showing that people with ancestral origins from different parts of Europe have different body measurements including height, with northern European ancestry associated with greater height and southern European ancestry associated with shorter height. This suggests that subcontinental differences in ancestry can have opposite effects on biological traits and diseases.
This finding suggests that the subcontinental differences in ancestry between individuals can have opposite effects on biological traits, diseases, and health outcomes, emphasizing the importance of not classifying individuals into broad ancestry groups such as African, European, or Asian. Doing this will help to make this research more accurate and will help to improve the field of precision medicine.
Researchers at Princeton University and the Simons Foundation have identified four clinically and biologically distinct subtypes of autism, marking a transformative step in understanding the condition’s genetic underpinnings and potential for personalised care.
Analysing data from over 5000 children in SPARK, an autism cohort study funded by the Simons Foundation, the researchers used a computational model to group individuals based on their combinations of traits. The team used a “person-centred” approach that considered a broad range of over 230 traits in each individual, from social interactions to repetitive behaviours to developmental milestones, rather than searching for genetic links to single traits.
This approach enabled the discovery of clinically relevant autism subtypes, which the researchers linked to distinct genetic profiles and developmental trajectories, offering new insights into the biology underlying autism. Their results were published July 9 in Nature Genetics.
The study defines four subtypes of autism: Social and Behavioural Challenges, Mixed ASD with Developmental Delay, Moderate Challenges, and Broadly Affected. Each subtype exhibits distinct developmental, medical, behavioural and psychiatric traits, and importantly, different patterns of genetic variation.
Individuals in the Social and Behavioural Challenges group show core autism traits, including social challenges and repetitive behaviours, but generally reach developmental milestones at a pace similar to children without autism. They also often experience co-occurring conditions like ADHD, anxiety, depression or obsessive-compulsive disorder alongside autism. One of the larger groups, this constitutes around 37% of the participants in the study.
The Mixed ASD with Developmental Delay group tends to reach developmental milestones, such as walking and talking, later than children without autism, but usually does not show signs of anxiety, depression or disruptive behaviours. “Mixed” refers to differences within this group with respect to repetitive behaviours and social challenges. This group represents approximately 19% of the participants.
Individuals with Moderate Challenges show core autism-related behaviours, but less strongly than those in the other groups, and usually reach developmental milestones on a similar track to those without autism. They generally do not experience co-occurring psychiatric conditions. Roughly 34% of participants fall into this category.
The Broadly Affected group faces more extreme and wide-ranging challenges, including developmental delays, social and communication difficulties, repetitive behaviours and co-occurring psychiatric conditions like anxiety, depression and mood dysregulation. This is the smallest group, accounting for around 10% of the participants.
“These findings are powerful because the classes represent different clinical presentations and outcomes, and critically we were able to connect them to distinct underlying biology,” said Aviya Litman, a PhD student at Princeton.
Distinct genetics behind the subtypes
For decades, autism researchers and clinicians have been seeking robust definitions of autism subtypes to aid in diagnosis and care. Autism is known to be highly heritable, with many implicated genes.
“While genetic testing is already part of the standard of care for people diagnosed with autism, thus far, this testing reveals variants that explain the autism of only about 20% of patients,” said co-author Jennifer Foss-Feig, a clinical psychologist at the Icahn School of Medicine at Mount Sinai and vice president and senior scientific officer at the Simons Foundation Autism Research Initiative (SFARI). This study takes an approach that differs from classic gene discovery efforts by identifying robust autism subtypes that are linked to distinct types of genetic mutations and affected biological pathways.
For example, children in the Broadly Affected group showed the highest proportion of damaging de novo mutations, while only the Mixed ASD with Developmental Delay group was more likely to carry rare inherited genetic variants. While children in both of these subtypes share some important traits like developmental delays and intellectual disability, these genetic differences suggest distinct mechanisms behind superficially similar clinical presentations.
“These findings point to specific hypotheses linking various pathways to different presentations of autism,” said Litman, referring to differences in biology between children with different autism subtypes.
Moreover, the researchers identified divergent biological processes affected in each subtype. “What we’re seeing is not just one biological story of autism, but multiple distinct narratives,” said Natalie Sauerwald, associate research scientist at the Flatiron Institute and co-lead author. “This helps explain why past genetic studies often fell short – it was like trying to solve a jigsaw puzzle without realising we were actually looking at multiple different puzzles mixed together. We couldn’t see the full picture, the genetic patterns, until we first separated individuals into subtypes.”
Autism biology unfolds on different timelines
The team also found that autism subtypes differ in the timing of genetic disruptions’ effects on brain development. Genes switch on and off at specific times, guiding different stages of development. While much of the genetic impact of autism was thought to occur before birth, in the Social and Behavioural Challenges subtype – which typically has substantial social and psychiatric challenges, no developmental delays, and a later diagnosis – mutations were found in genes that become active later in childhood. This suggests that, for these children, the biological mechanisms of autism may emerge after birth, aligning with their later clinical presentation.
“By integrating genetic and clinical data at scale, we can now begin to map the trajectory of autism from biological mechanisms to clinical presentation,” said co-author Chandra Theesfeld, senior academic research manager at the Lewis-Sigler Institute and Princeton Precision Health.
A paradigm shift for autism research
This study builds on more than a decade of autism genomics research led by Troyanskaya and collaborators. It is enabled by the close integration of interdisciplinary expertise in genomics, clinical psychology, molecular biology, computer science and modelling, and computational biology.
“The Princeton Precision Health initiative uses artificial intelligence and computational modelling to integrate across biological and clinical data,” said Jennifer Rexford, Princeton University provost and Gordon Y.S. Wu Professor in Engineering. “This initiative could not exist without the University’s charitable endowment. Our investments allow experts to collaborate across a range of disciplines to conduct transformative research that improves human health, including the potential for major advances in the diagnosis and treatment of autism made possible in this exciting project.”
“It’s a whole new paradigm, to provide these groups as a starting point for investigating the genetics of autism,” said Theesfeld. Instead of searching for a biological explanation that encompasses all individuals with autism, researchers can now investigate the distinct genetic and biological processes driving each subtype.
This shift could reshape both autism research and clinical care – helping clinicians anticipate different trajectories in diagnosis, development and treatment. “The ability to define biologically meaningful autism subtypes is foundational to realising the vision of precision medicine for neurodevelopmental conditions,” said Sauerwald.
While the current work defines four subtypes, “this doesn’t mean there are only four classes,” said Litman. “It means we now have a data-driven framework that shows there are at least four – and that they are meaningful in both the clinic and the genome.”
Looking ahead
Beyond its contributions to understanding autism subtypes and their underlying biology, the study offers a powerful framework for characterising other complex, heterogeneous conditions and finding clinically relevant disease subtypes. As Theesfeld put it: “This opens the door to countless new scientific and clinical discoveries.”
New USC research indicates how iron-related oxidative damage and cell death may hasten the development of Alzheimer’s disease in people with Down syndrome
Scientists at the University of Southern Carolina have discovered a key connection between high levels of iron in the brain and increased cell damage in people who have both Down syndrome and Alzheimer’s disease.
In the study, researchers found that the brains of people diagnosed with Down syndrome and Alzheimer’s disease (DSAD) had twice as much iron and more signs of oxidative damage in cell membranes compared to the brains of individuals with Alzheimer’s disease alone or those with neither diagnosis. The results, published in Alzheimer’s & Dementia: The Journal of the Alzheimer’s Association, point to a specific cellular death process that is mediated by iron, and the findings may help explain why Alzheimer’s symptoms often appear earlier and more severely in individuals with Down syndrome.
“This is a major clue that helps explain the unique and early changes we see in the brains of people with Down syndrome who develop Alzheimer’s,” said Max Thorwald, lead author of the study and a postdoctoral fellow in the laboratory of University Professor Emeritus Caleb Finch at the USC Leonard Davis School. “We’ve known for a long time that people with Down syndrome are more likely to develop Alzheimer’s disease, but now we’re beginning to understand how increased iron in the brain might be making things worse.”
Down syndrome and Alzheimer’s
Down syndrome is caused by having an extra third copy, or trisomy, of chromosome 21. This chromosome includes the gene for amyloid precursor protein, or APP, which is involved in the production of amyloid-beta (Aβ), the sticky protein that forms telltale plaques in the brains of people with Alzheimer’s disease.
Because people with Down syndrome have three copies of the APP gene instead of two, they tend to produce more of this protein. By the age of 60, about half of all people with Down syndrome show signs of Alzheimer’s disease, which is approximately 20 years earlier than in the general population.
“This makes understanding the biology of Down syndrome incredibly important for Alzheimer’s research,” said Finch, the study’s senior author.
Key findings point to ferroptosis
The research team studied donated brain tissue from individuals with Alzheimer’s, DSAD, and those without either diagnosis. They focused on the prefrontal cortex — an area of the brain involved in thinking, planning, and memory — and made several important discoveries:
Iron levels much higher in DSAD brains: Compared to the other groups, DSAD brains had twice the amount of iron in the prefrontal cortex. Scientists believe this buildup comes from tiny brain blood vessel leaks called microbleeds, which occur more frequently in DSAD than in Alzheimer’s and are correlated with higher amounts of APP.
More damage to lipid-rich cell membranes: Cell membranes are made of fatty compounds called lipids and can be easily damaged by chemical stress. In DSAD brains, the team found more byproducts of this type of damage, known as lipid peroxidation, compared to amounts in Alzheimer’s-only or control brains.
Weakened antioxidant defense systems: The team found that the activity of several key enzymes that protect the brain from oxidative damage and repair cell membranes was lower in DSAD brains, especially in areas of the cell membrane called lipid rafts.
Together, these findings indicate increased ferroptosis, a type of cell death characterised by iron-dependent lipid peroxidation, Thorwald explained: “Essentially, iron builds up, drives the oxidation that damages cell membranes, and overwhelms the cell’s ability to protect itself.”
Lipid rafts: a hotspot for brain changes
The researchers paid close attention to lipid rafts — tiny parts of the brain cell membrane that play crucial roles in cell signalling and regulate how proteins like APP are processed. They found that in DSAD brains, lipid rafts had much more oxidative damage and fewer protective enzymes compared to Alzheimer’s or healthy brains.
Notably, these lipid rafts also showed increased activity of the enzyme β-secretase, which interacts with APP to produce Aβ proteins. The combination of more damage and more Aβ production may promote the growth of amyloid plaques, thus speeding up Alzheimer’s progression in people with Down syndrome, Finch explained.
Rare Down syndrome variants offer insight
The researchers also studied rare cases of individuals with “mosaic” or “partial” Down syndrome, in which the third copy of chromosome 21 is only present in a smaller subset of the body’s cells. These individuals had lower levels of APP and iron in their brains and tended to live longer. In contrast, people with full trisomy 21 and DSAD had shorter lifespans and higher levels of brain damage.
“These cases really support the idea that the amount of APP — and the iron that comes with it — matters a lot in how the disease progresses,” Finch said.
Looking ahead
The team says their findings could help guide future treatments, especially for people with Down syndrome who are at high risk of Alzheimer’s. Early research in mice suggests that iron-chelating treatments, in which medicine binds to the metal ions and allows them to leave the body, may reduce indicators of Alzheimer’s pathology, Thorwald noted.
“Medications that remove iron from the brain or help strengthen antioxidant systems might offer new hope,” Thorwald said. “We’re now seeing how important it is to treat not just the amyloid plaques themselves but also the factors that may be hastening the development of those plaques.”
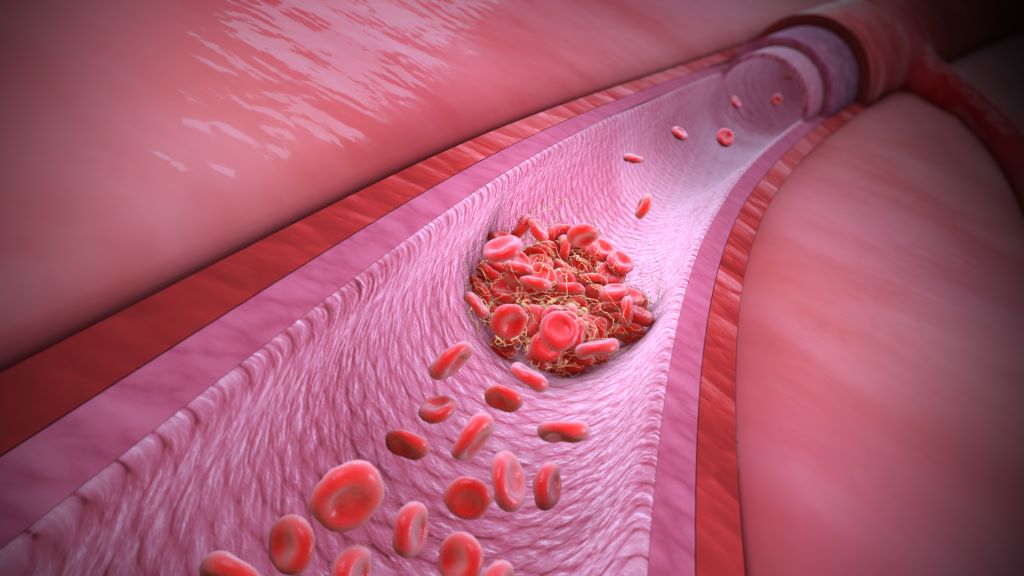
Though blood clots can form in both arteries and veins, the reasons behind them differ, as do the consequences and the chances of preventing blood clots. In Sweden, almost half of all cases of venous thrombosis have a genetic explanation. A team of researchers from Lund University in Sweden has now discovered three gene variants that increase the risk of blood clots in the leg by up to 180%.
There is a difference between arterial and venous blood clots. Blood clots in the arteries form when plaque in calcified vessels bursts and the body perceives it as an injury. This activates the platelets, which clump together and form a clot. In the worst case, it can lead to a stroke or heart attack. A venous thrombus, on the other hand, usually forms in the leg when the blood stagnates for too long. This can activate the body’s coagulation system, allowing the clotting system to be activated and the blood to clot, blocking blood flow. If the clot breaks loose and travels with the blood to the lungs, it can lead to pulmonary embolism, a life-threatening condition.
“Venous thrombosis is in fact one of the most common causes of death in the world. It is a common disease that has always been somewhat overshadowed by arterial blood clots,” says Bengt Zöller, a specialist in general medicine at Skåne University Hospital and professor of general medicine at Lund University.
In Sweden, more than 10 000 people suffer from venous thromboembolism each year and that number appears to be increasing. Several factors are contributing to this increase. One of the strongest risk factors is age, and as the number of older people in Sweden grows, the number of clots is also increasing. Ten per cent of 80-year-olds experience a blood clot at some point. The risk also increases if you are overweight or tall.
“The muscles control the blood flow in the veins and the legs become like columns of fluid where the force of gravity is strong. Too much sedentary and inactive behaviour, then, is harmful. Only the valves of the veins prevent backflow and if these are damaged, the risk of blood clots can increase. Therefore, tall people are more prone to blood clots, as their larger veins provide less blood flow, combined with the fact that blood must travel a greater distance back to the heart.”
Because the heart pumps blood out into the arteries, there is much higher blood pressure in the arteries than in the veins, which can contribute to atherosclerosis. High blood pressure, high levels of blood lipids and smoking are all risk factors for atherosclerosis of the arteries. But because the veins are a low-pressure system, the vessels do not become atherosclerotic. Therefore, neither high blood pressure nor blood lipids are associated with venous clots and smoking is considered only a weak to moderate risk factor. Being overweight, on the other hand, is a very significant culprit. Obesity has a negative impact on venous circulation, especially when combined with the fact that overweight people are often less active. Some clotting factors are also affected by obesity.
“In terms of diet, there are fewer studies, but ultra-processed foods have been associated with a slightly increased risk of blood clots, and plant-based, healthy foods with a reduced risk. In our studies, we have also seen that commercial fishermen have a lower risk, which may be due to a higher omega-3 content in their diet.”
There are also specific situations in which the risk of venous blood clots is particularly high. The risk of blood clots increases when blood flow is reduced, such as when travelling by air for long periods of time or when lying in bed for several days. Surgery or inflammation that damages the vessel wall can also lead to an increased tendency to clot. Particularly during pregnancy, blood clotting factors increase and levels of some protective proteins may decrease.
“In these risk situations, prophylaxis in the form of blood thinners may be particularly important if other risk factors are also present.”
Other risk factors are the genetic variants that affect different parts of the blood’s clotting ability. In Sweden, we have a high prevalence of APC (activated protein C) resistance due to an inherited mutation in the gene for coagulation factor V, called Factor V Leiden. About 10 per cent of Swedes have this mutation, which is considered the most common coagulation mutation among Indo-Europeans.
“Evolutionarily, bleeding less has been an advantage, but in our modern, sedentary society, APC resistance is becoming a risk factor.”
Bengt Zöller and his fellow researchers have now identified the strongest genetic risk factor since Factor V Leiden was discovered. They used data from the population-based Malmö Kost Cancer study, involving 30,000 Malmö residents. By selecting 27 genes previously associated with clotting disorders, they found three variants that, when taken together, were as significant a risk factor for venous blood clots as Factor V Leiden: ABO, F8, and VWF each increased the risk of venous blood clots by 10 to 30 percent.
“And the more of these variants a person has – the higher the risk. An individual with five of these gene variants has a 180 per cent higher risk of venous thrombosis. Unlike Factor V Leiden, which is only found in Indo-Europeans, these three different mutations are found in between five and fifty per cent of various populations around the globe.”
As these genetic variants are present in all populations, the next step is to investigate how the number of risk genes affects the duration of treatment with anticoagulants after a blood clot.
“I think tailoring treatment based on risk assessment will become increasingly important,” concludes Bengt Zöller.
Genetic ancestry is much more complicated than how people report their race and ethnicity. New research, using data from the National Institutes of Health’s (NIH) All of Us Research Program, finds that people who identify as being from the same race or ethnic group can have a wide range of genetic differences. The findings are reported June 5 in the American Journal of Human Genetics.
As doctors and researchers learn more about how genetic variants influence the incidence and course of human diseases, the study of genetic ancestry has become increasingly important. This research is driving the field of precision medicine, which aims to develop individualized healthcare.
People whose ancestors came from the same part of the world are likely to have inherited the same genetic variants, but self-identified race and ethnicity don’t tell the whole story about a person’s ancestors. NIH’s All of Us Research Program was created in part to address this puzzle and to learn more about how genetic ancestry influences human health.
In the current study, the investigators looked at the DNA of more than 230 000 people who have volunteered to share their health information for All of Us. They compared it to other large DNA projects from around the world using a technique called principal component analysis (PCA) to visualise population structure and help identify genetic similarity between individuals and groups of people. This analysis showed that people in the US have very mixed ancestry, and their DNA doesn’t always match the race or ethnicity they write on forms. Instead of falling into clear groups based on race or ethnicity, people’s genetic backgrounds show gradients of variation across different US regions and states.
This is especially significant for people who identify as being of Hispanic or Latino origin. These people have a wide-ranging blend of ancestries from European, Native American, and African groups. Importantly, genetic ancestry among these people varies across the US in part because of historic migration patterns. For example, Hispanics/Latinos in the Northeast are more likely to have Caribbean (and thus African) ancestry, and those in the Southwest are more likely to have Mexican and Central American (and thus Native American) ancestry.
One specific discovery was that ancestry was significantly associated with body mass index (BMI) and height, even after adjusting for socio-economic differences. For example, West and Central African ancestries were associated with higher BMI, whereas East Africa ancestry was associated with lower BMI. There were similar findings showing that people with ancestral origins from different parts of Europe have different body measurements including height, with northern European ancestry associated with greater height and southern European ancestry associated with shorter height. This suggests that subcontinental differences in ancestry can have opposite effects on biological traits and diseases.
This finding suggests that the subcontinental differences in ancestry between individuals can have opposite effects on biological traits, diseases, and health outcomes, emphasising the importance of not classifying individuals into broad ancestry groups such as African, European, or Asian. Doing this will help to make this research more accurate and will help to improve the field of precision medicine.
New research has found that men who carry a common genetic variant are twice as likely to develop dementia in their lifetime compared to women. The research, published in Neurology, used data from the ASPirin in Reducing Events in the Elderly (ASPREE) trial to investigate whether people who had variants in the haemochromatosis (HFE) gene, which is critical for regulating iron levels in the body, might be at increased risk of dementia.
Co-author Professor John Olynyk, from the Curtin Medical School, said one in three people carry one copy of the variant, known as H63D, while one in 36 carry two copies.
“Having just one copy of this gene variant does not impact someone’s health or increase their risk of dementia. However, having two copies of the variant more than doubled the risk of dementia in men, but not women,” Professor Olynyk said.
“While the genetic variant itself cannot be changed, the brain pathways which it affects – leading to the damage that causes dementia – could potentially be treated if we understood more about it.”
Professor Olynyk said further research was needed to investigate why this genetic variant increased the risk of dementia for males but not females.
“The HFE gene is routinely tested for in most Western countries including Australia when assessing people for haemochromatosis – a disorder that causes the body to absorb too much iron. Our findings suggest that perhaps this testing could be offered to men more broadly,” Professor Olynyk said.
“While the HFE gene is critical for controlling iron levels in the body, we found no direct link between iron levels in the blood and increased dementia risk in affected men.
“This points to other mechanisms at play, possibly involving the increased risk of brain injury from inflammation and cell damage in the body.”
The ASPREE trial was a double-blind, randomised, placebo-controlled trial of daily low-aspirin in 19 114 healthy older people in Australia and the USA. Primarily undertaken to evaluate the risks versus benefits of daily low-dose aspirin in this cohort, it created a treasure trove of healthy ageing data that has underpinned a wealth of research studies.