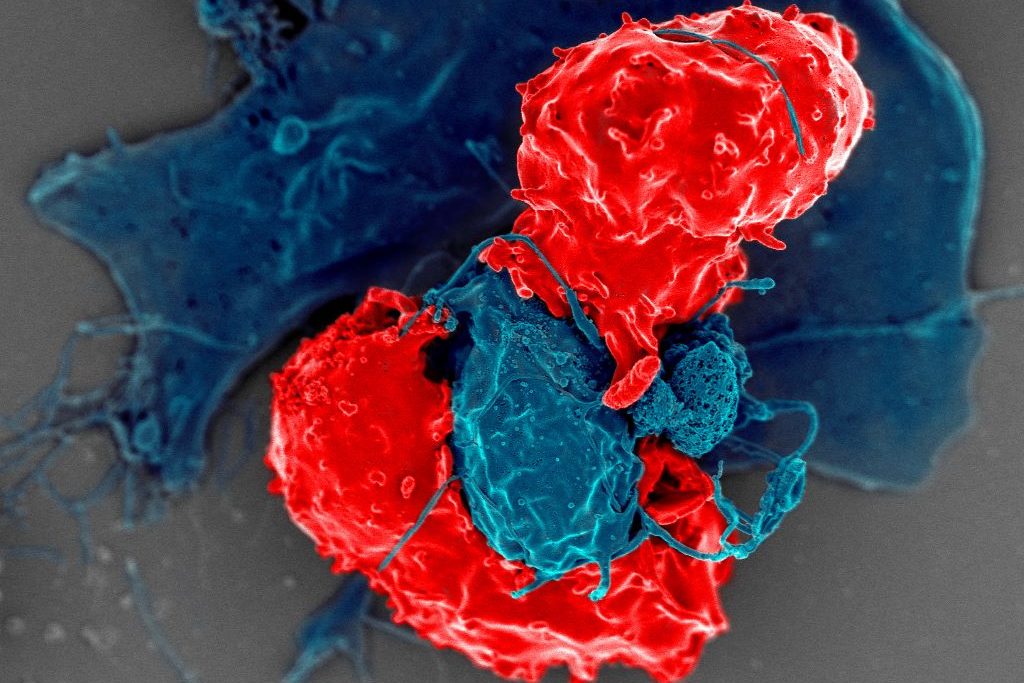
Scanning electron microscope image of T regulatory cells (red) interacting with antigen-presenting cells (blue). T regulatory cells can suppress responses by T cells to maintain homeostasis in the immune system. Credit: National Institute of Allergy and Infectious Diseases/NIH
Cedars-Sinai Health Sciences University investigators have identified for the first time a protein’s role as a “dimmer switch” that can calm an overactive immune system and restrain harmful inflammation. The protein, Butyrophilin 2A2 (BTN2A2), interacts with a key molecule that controls the strength of T-cell responses.
The findings, published inNature Communications, define a unique pathway that helps balance immune activity and could be harnessed to limit damage caused by a variety of autoimmune diseases.
In laboratory mice, loss of BTN2A2 led to exaggerated immune reactions and an increase in damaging kidney inflammation called glomerulonephritis. Treatment with BTN2A2 reduced disease severity by increasing immune-regulating T cells and lowering inflammation.
Supporting laboratory experiments in human T-cells demonstrated similar immune-calming effects.
“Glomerulonephritis remains a leading cause of chronic kidney disease and kidney failure worldwide, with limited treatment options,” said Ananth Karumanchi, MD, co-corresponding author of the study and director of the Renovascular Research Center at Cedars-Sinai. “Our findings provide a strong foundation for future studies aimed at modifying immune-driven kidney disease rather than simply managing its symptoms. The pathway could also be targeted in a range of autoimmune and inflammatory diseases including rheumatoid arthritis, multiple sclerosis, inflammatory bowel disease, and transplant rejections.”
Other Cedars-Sinai authors include Shafat Ali, Anders H. Berg, Michifumi Yamashita, Ambart E. Covarrubias, Jordan Mundell, Pranali N. Shah, Ruan Zhang, Vincent Dupont, Bong-Ha Shin, Shen Yang, Madhusudhanarao Katiki, Ramachandran Murali, Margareta D. Pisarska, Ravi Thadhani, Peter S. Heeger and Stanley C. Jordan
A new ACC Scientific Statement on Inflammation and Cardiovascular Disease (CVD) highlights new groundbreaking research linking inflammation atherosclerotic cardiovascular disease (ASCVD) and provides consensus-based recommendations for evaluation, treatment and prevention reflecting this new era.
“The evidence linking inflammation with ASCVD is no longer exploratory but is compelling and clinically actionable,” write the authors, led by Writing Committee Chair George A. Mensah, MD, FACC. “The time for taking action has now arrived.”
Published in JACC, the Statement includes specific recommendations for screening, evaluation, and CVD risk assessment; inflammatory biomarkers in cardiovascular imaging; inflammation inhibition in behavioural and lifestyle risks; and anti-inflammatory approaches in primary and secondary prevention, as well as in heart failure and other cardiovascular diseases.
Among the key takeaways:
High-sensitivity C-reactive protein (hsCRP) is an inexpensive and widely available blood test. While there has been debate within the medical community regarding the utility of hsCRP, this statement details the data confirming its value in clinical decision making in primary and secondary prevention.
In patients with known CVD, hsCRP level is at least as predictive of future events as LDL cholesterol levels, even in patients treated with statin therapy.
The important role of lifestyle interventions to reduce systemic inflammation is emphasised, including regular exercise (at least 150 minutes/week), Mediterranean or DASH Diet, and intake of omega-3 fatty acids, including two to three meals per week of fatty fish high in EPA and DHA. This advice aligns with lifestyle management recommendations in the 2025 ACC/AHA High Blood Pressure Guideline
The Statement also discusses current challenges and opportunities based on the new evidence, exploring topics like the advancing field of cardioimmunology and areas for further research, such as the interplay between inflammation and key physiological systems, the role of novel special pro-resolving bioactive lipid molecules in promoting the resolution of inflammation and CVD risk reduction, and more.
The authors close with a call for action to “embrace anti-inflammatory interventions in patients with established ASCVD” and for clinical practice guidelines that implement “broad screening of primary and secondary prevention patients for hsCRP, in combination with LDL cholesterol.” Additionally, they note: “The time is also ripe for the development of strategies to promote increased physician awareness of the crucial role of inflammation in CVD and accelerate the adoption of evidence-based, guideline-directed anti-inflammatory therapy through dissemination and implementation research.”
Cardiologists have long known that up to half of all heart attacks and strokes occur among apparently healthy individuals who do not smoke and do not have high blood pressure, high cholesterol, or diabetes, the “standard modifiable risk factors” which doctors often call “SMuRFs.” How to identify risk among the “SMuRF-Less” has been an elusive goal in preventive cardiology, particularly in women who are often under-diagnosed and under-treated.
A new study by Mass General Brigham researchers that leverages data from the Women’s Health Study has found hsCRP, a marker of inflammation, can help identify women who are at risk but are missed by current screening algorithms. Results are presented at a late-breaking clinical science session at the European Society of Cardiology Congress (ESC) and simultaneously published in The European Heart Journal.
“Women who suffer from heart attacks and strokes yet have no standard modifiable risk factors are not identified by the risk equations doctors use in daily practice,” said Paul Ridker, MD, MPH, a preventive cardiologist at Mass General Brigham’s Heart and Vascular Institute. “Yet our data clearly show that apparently healthy women who are inflamed are at substantial lifetime risk. We should be identifying these women in their 40s, at a time when they can initiate preventive care, not wait for the disease to establish itself in their 70s when it is often too late to make a real difference.”
As part of the federally funded study, researchers studied 12 530 initially healthy women with no standard modifiable risk factors who had the inflammatory biomarker hsCRP measured at study entry and who were then followed over 30 years. Despite the lack of traditional risks, women who were inflamed as defined by hsCRP levels > 3 mg/L had a 77% increased lifetime risk of coronary heart disease, a 39% increased lifetime risk of stroke, and a 52% increased lifetime risk of any major cardiovascular event.
Additionally, researchers released a new analysis of randomised trial data showing that “SMuRF-Less but Inflamed” patients can reduce their risk of heart attack and stroke by 38% using statin therapy.
“While those with inflammation should aggressively initiate lifestyle and behavioural preventive efforts, statin therapy could also play an important role in helping reduce risk among these individuals,” said Ridker.
A woman with Systemic Lupus Erythematosus. Source: Wikimedia CC0
Chronic inflammation occurs when the immune system is stuck in attack mode, sending cell after cell to defend and repair the body for months or even years. Diseases associated with chronic inflammation, like arthritis or cancer or autoimmune disorders, weigh heavily on human health – and their incidence is expected to rise. A new study by investigators from Mass General Brigham identified a protein called WSTF that could be targeted to block chronic inflammation. Crucially, this strategy would not interfere with acute inflammation, allowing the immune system to continue responding appropriately to short-term threats, such as infection by a pathogen. Results are published in Nature.
“Chronic inflammatory diseases cause a great deal of suffering and death, but we still have much to learn about what drives chronic inflammation and how to treat it,” said senior author Zhixun Dou, PhD, of the Center for Regenerative Medicine and Krantz Family Center for Cancer Research at Massachusetts General Hospital. “Our findings help us separate chronic and acute inflammation, as well as identify a new target for stopping chronic inflammation that results from aging and disease.”
Using chronically inflamed human cells, the researchers found that WSTF interacts with other proteins inside cell nuclei, which prompts its excretion and degradation. Since WSTF is responsible for concealing pro-inflammatory genes, this nucleus-eviction reveals those genes and, in turn, amplifies inflammation. They confirmed that WSTF loss could promote inflammation in mouse models of aging and cancer. They also found, using human cells, that WSTF loss only occurred in chronic inflammation, not acute. Using these findings, the researchers designed a WSTF-restoring therapeutic to suppress chronic inflammation and observed preliminary success in mouse models of aging, metabolic dysfunction-associated steatohepatitis (MASH), and osteoarthritis.
The researchers went further to examine tissue samples from patients with MASH or osteoarthritis. They found that WSTF is lost in the livers of patients with MASH, but not in the livers of healthy donors. Using cells from the knees of osteoarthritis patients undergoing joint replacement surgery, they showed that WSTF-restoring therapeutic reduces chronic inflammation from the inflamed knee cells. These findings highlight the potential of developing new treatments targeting WSTF to combat chronic inflammatory diseases.
Further research is needed to validate the therapeutic potential of WSTF restoration in broader settings and to develop specific strategies to target WSTF. Additionally, the findings suggest other similar proteins may be involved in chronic inflammation, opening a promising new avenue for studying and treating inflammation in the future.
Researchers at UCLA Health uncovered new information about the role inflammation plays in mitigating liver fibrosis, which is associated with metabolic-associated fatty liver disease (MAFLD). While inflammation in the liver has long been considered a prerequisite to developing liver fibrosis, the scarring and thickening of tissue that can impair the liver’s ability to function, this new research, published in the Journal of Clinical Investigation, suggests that reducing inflammation may not influence the extent of fibrosis.
“Liver fibrosis is the critical feature that creates chronic liver disease and liver cancer. If we can keep fibrosis in check then we can meaningfully impact liver disease,” said Tamer Sallam, MD, corresponding author of the study and vice chair and associate professor in the department of medicine at the David Geffen School of Medicine at UCLA.
“For decades we have believed that targeting inflammation is one of the most important ways to reduce MAFLD. But this new research indicates that inflammation, while still important, may not be the main driver of fibrosis.”
The studylooked specifically at a protein called lipopolysaccharide binding protein (LBP), which is involved in the body’s immune response, and how LBP functions in mice. Findings showed that mice without LBP in their liver cells had lower levels of liver inflammation and better liver function but no change in fibrosis.
In addition to mouse models, the researchers also studied genetic analyses from large human datasets and human tissue samples from MAFLD patients at different stages in the disease, to examine the consequence of loss of LBP function. The evidence combined showed that the LBP does not alter scar tissue markers.
Sallam indicated a need to further explore how LBP influences inflammation and whether other factors can offer a more potent reduction in inflammation and have an impact on reducing fibrosis.
“Reducing scar burden is one of the holy grails in the treatment of advanced liver diseases,” Sallam said. “These results suggest that certain ways of targeting inflammation may not be a viable option and that more directed therapies against other pathways could help us better target fibrosis and improve outcomes for patients.”
Infections and neurodegenerative diseases cause inflammation in the brain. But for unknown reasons, patients with brain inflammation often develop muscle problems that seem to be independent of the central nervous system. Now, researchers at Washington University School of Medicine in St. Louis have revealed how brain inflammation releases a specific protein that travels from the brain to the muscles and causes a loss of muscle function.
The study, published in Science Immunology, also identified ways to block this process, which could have implications for treating or preventing the muscle wasting sometimes associated with inflammatory diseases, including bacterial infections, Alzheimer’s disease and long COVID.
“We are interested in understanding the very deep muscle fatigue that is associated with some common illnesses,” said senior author Aaron Johnson, PhD, an associate professor of developmental biology. “Our study suggests that when we get sick, messenger proteins from the brain travel through the bloodstream and reduce energy levels in skeletal muscle. This is more than a lack of motivation to move because we don’t feel well. These processes reduce energy levels in skeletal muscle, decreasing the capacity to move and function normally.”
Fruit fly and mouse models
To investigate the effects of brain inflammation on muscle function, the researchers modelled three different types of diseases – an E. coli bacterial infection, a SARS-CoV-2 viral infection and Alzheimer’s. When the brain is exposed to inflammatory proteins characteristic of these diseases, damaging chemicals called reactive oxygen species build up. The reactive oxygen species cause brain cells to produce an immune-related molecule called interleukin-6 (IL-6), which travels throughout the body via the bloodstream. The researchers found that IL-6 in mice – and the corresponding protein in fruit flies – reduced energy production in muscles’ mitochondria, the energy factories of cells.
“Flies and mice that had COVID-associated proteins in the brain showed reduced motor function – the flies didn’t climb as well as they should have, and the mice didn’t run as well or as much as control mice,” Johnson said. “We saw similar effects on muscle function when the brain was exposed to bacterial-associated proteins and the Alzheimer’s protein amyloid beta. We also see evidence that this effect can become chronic. Even if an infection is cleared quickly, the reduced muscle performance remains many days longer in our experiments.”
Johnson, along with collaborators at the University of Florida and first author Shuo Yang, PhD (who did this work as a postdoctoral researcher in Johnson’s lab) make the case that the same processes are likely relevant in people. The bacterial brain infection meningitis is known to increase IL-6 levels and can be associated with muscle issues in some patients, for instance. Among COVID-19 patients, inflammatory SARS-CoV-2 proteins have been found in the brain during autopsy, and many long COVID patients report extreme fatigue and muscle weakness even long after the initial infection has cleared. Patients with Alzheimer’s disease also show increased levels of IL-6 in the blood as well as muscle weakness.
Potential treatment targets
The study pinpoints potential targets for preventing or treating muscle weakness related to brain inflammation. The researchers found that IL-6 activates what is called the JAK-STAT pathway in muscle, and this is what causes the reduced energy production of mitochondria. Several therapeutics already approved by the Food and Drug Administration for other diseases can block this pathway. JAK inhibitors as well as several monoclonal antibodies against IL-6 are approved to treat various types of arthritis and manage other inflammatory conditions.
“We’re not sure why the brain produces a protein signal that is so damaging to muscle function across so many different disease categories,” Johnson said. “If we want to speculate about possible reasons this process has stayed with us over the course of human evolution, despite the damage it does, it could be a way for the brain to reallocate resources to itself as it fights off disease. We need more research to better understand this process and its consequences throughout the body.
“In the meantime, we hope our study encourages more clinical research into this pathway and whether existing treatments that block various parts of it can help the many patients who experience this type of debilitating muscle fatigue,” he said.
Scientists at King’s College London have discovered that the features of asthma attacks, a disease usually treated as being inflammatory, in fact stem from constriction of airways, making breathing difficult. The new study, published in Science, shows for the first time that many features of an asthma attack – inflammation, mucus secretion, and damage to the airway barrier that prevents infections – result from this mechanical constriction in a mouse model.
The findings suggest that blocking a process that normally causes epithelial cell death could prevent the damage, inflammation, and mucus that result from an asthma attack.
Professor Jody Rosenblatt from King’s College London said: “Our discovery is the culmination of more than ten years work. As cell biologists who watch processes, we could see that the physical constriction of an asthma attack causes widespread destruction of the airway barrier. Without this barrier, asthma sufferers are far more likely to get long-term inflammation, wound healing, and infections that cause more attacks. By understanding this fundamental mechanism, we are now in a better position to prevent all these events.”
Asthma symptoms include wheezing, coughing, feeling breathlessness and a tight chest. Triggers such as pollen or dust can make asthma symptoms worse and can lead to a life-threatening asthma attack.
Despite the disease commonality, the causes of asthma are still not understood. Current medications treat the consequences of an asthma attack by opening the airways, calming inflammation, and breaking up the sticky mucus which clogs the airway, which help control asthma, but do not prevent it.
The answer to stopping asthma symptoms may lie in cell extrusion, a process the researchers discovered that drives most epithelial cell death. Scientists used mouse lung models and human airway tissue to discover that when the airways contract, known as bronchoconstriction, the epithelial cells that line the airway get squeezed out to later die.
Because bronchoconstriction causes so many cell extrusions, it damages the airway barrier which causes inflammation and excess mucus.
In previous studies, the scientists found that the chemical compound gadolinium can block extrusion. In this study, they found it could work in mice to prevent the excess extrusion that causes damage and inflammation after an asthma attack. The authors note that gadolinium has not been tested in humans and has not been deemed to be safe or efficacious.
Professor Rosenblatt said: “This constriction and destruction of the airways causes the post-attack inflammation and excess mucus secretion that makes it difficult for people with asthma to breathe.
“Current therapies do not prevent this destruction – an inhaler such as Albuterol opens the airways, which is critical to breathing but, dishearteningly, we found it does not prevent the damage and the symptoms that follow an attack. Fortunately, we found that we can use an inexpensive compound, gadolinium which is frequently used for MRI imaging, to stop the airway damage in mice models as well as the ensuing inflammation and mucus secretion. Preventing this damage could then prevent the build-up of musculature that cause future attacks.”
Professor Chris Brightling from the University of Leicester and one of the co-authors of the study said: “In the last decade there has been tremendous progress in therapies for asthma particularly directed towards airway inflammation. However, there remains ongoing symptoms and attacks in many people with asthma. This study identifies a new process known as epithelial extrusion whereby damage to the lining of the airway occurs as a consequence of mechanical constriction and can drive many of the key features of asthma. Better understanding of this process is likely to lead to new therapies for asthma.”
The discovery of the mechanics behind cell extrusion could underlie other inflammatory diseases that also feature constriction such as cramping of the gut and inflammatory bowel disease.
Just as you can’t make an omelette without breaking eggs, scientists at Albert Einstein College of Medicine have found that you can’t make long-term memories without DNA damage and inflammation in the brain. Their surprising findings were published online today in the journal Nature.
“Inflammation of brain neurons is usually considered to be a bad thing, since it can lead to neurological problems such as Alzheimer’s and Parkinson’s disease,” said study leader Jelena Radulovic, MD, PhD, professor of psychiatry and behavioural sciences at Einstein. “But our findings suggest that inflammation in certain neurons in the brain’s hippocampal region is essential for making long-lasting memories.”
The hippocampus has long been known as the brain’s memory centre. Dr Radulovic and her colleagues found that a stimulus sets off a cycle of DNA damage and repair within certain hippocampal neurons that leads to stable memory assemblies, ie clusters of brain cells representing past experiences.
From shocks to stable memories
The researchers discovered this memory-forming mechanism by giving mice brief, mild shocks sufficient to form an episodic memory of the shock event. Then, they analysed neurons in the hippocampal region and found that genes participating in an important inflammatory signalling pathway had been activated.
“We observed strong activation of genes involved in the Toll-Like Receptor 9 (TLR9) pathway,” said Dr Radulovic, who is also director of the Psychiatry Research Institute at Montefiore Einstein (PRIME). “This inflammatory pathway is best known for triggering immune responses by detecting small fragments of pathogen DNA. So at first we assumed the TLR9 pathway was activated because the mice had an infection. But looking more closely, we found, to our surprise, that TLR9 was activated only in clusters of hippocampal cells that showed DNA damage.”
Brain activity routinely induces small breaks in DNA that are repaired within minutes. But in this population of hippocampal neurons, the DNA damage appeared to be more substantial and sustained.
Triggering inflammation to make memories
Further analysis showed that DNA fragments, along with other molecules resulting from the DNA damage, were released from the nucleus, after which the neurons’ TLR9 inflammatory pathway was activated; this pathway in turn stimulated DNA repair complexes to form at an unusual location: the centrosomes. These organelles are present in the cytoplasm of most animal cells and are essential for coordinating cell division. But in neurons – which don’t divide – the stimulated centrosomes participated in cycles of DNA repair that appeared to organise individual neurons into memory assemblies.
“Cell division and the immune response have been highly conserved in animal life over millions of years, enabling life to continue while providing protection from foreign pathogens,” Dr. Radulovic said. “It seems likely that over the course of evolution, hippocampal neurons have adopted this immune-based memory mechanism by combining the immune response’s DNA-sensing TLR9 pathway with a DNA repair centrosome function to form memories without progressing to cell division.”
Resisting inputs of extraneous information
During the week required to complete the inflammatory process, the mouse memory-encoding neurons were found to have changed in various ways, including becoming more resistant to new or similar environmental stimuli. “This is noteworthy,” said Dr Radulovic, “because we’re constantly flooded by information, and the neurons that encode memories need to preserve the information they’ve already acquired and not be ‘distracted’ by new inputs.”
“This is noteworthy,” said Dr Radulovic, “because we’re constantly flooded by information, and the neurons that encode memories need to preserve the information they’ve already acquired and not be ‘distracted’ by new inputs.”
Importantly, the researchers found that blocking the TLR9 inflammatory pathway in hippocampal neurons not only prevented mice from forming long-term memories but also caused profound genomic instability, ie, a high frequency of DNA damage in these neurons.
“Genomic instability is considered a hallmark of accelerated aging as well as cancer and psychiatric and neurodegenerative disorders such as Alzheimer’s,” Dr Radulovic said.
“Drugs that inhibit the TLR9 pathway have been proposed for relieving the symptoms of long COVID. But caution needs to be shown because fully inhibiting the TLR9 pathway may pose significant health risks.”
PhD Student Elizabeth Wood and Ana Cicvaric, a postdoc in the Radulovic lab, were the study’s first authors at Einstein.
In the human body, a protein carrier called SPNS2 transports S1P molecules from endothelial cells to rally immune cell response in infected organs and tissues, resulting in inflammation. By enlarging the entire SPNS2 structure using nanoparticles, the S1P molecules contained within can be viewed via cryogenic electron microscopy. Using this information, small molecules can be developed to inhibit this signalling pathway and treat inflammatory diseases.
Scientists at the National University of Singapore and colleagues in China have analysed the structure of the SPNS2 protein at an atomic level that could provide greater insights into how S1P signalling molecules are released to communicate with the immune cells to regulate inflammatory responses. Their findings are published in Cell Research.
“Seeing is believing. This work shows that SPNS2 is directly exporting S1P for signalling and it is possible to inhibit its transport function with small molecules. This work provides the foundation for understanding of how S1P is released by SPNS2 and how this protein function is inhibited by small molecules for treatment of inflammatory diseases,” said team leader Dr Nguyen Nam Long.
The SPNS2 protein allows the binding of the S1P signalling molecules to trigger the immune cells to leave the lymph nodes and induce inflammation in different parts of the body when needed.
Made up of amino acids, the SPNS2 protein is malleable enough to change its shape and structure to release the S1P signalling molecules through small cavities found within the protein.
Through the discovery of how the SPNS2 protein releases S1P molecules, the SPNS2 structure can be exploited for future drug development.
Similar to discovering how the shape of the lock looks like before the key can be designed, this finding sheds more light into how future drugs can be designed to better target the protein to increase drug efficacy.
This finding builds on previous research which found that deleting SPNS2 protein from a pre-clinical model effectively blocks the S1P signalling pathway so that the S1P signalling molecules are unable to be transported to prompt immune cells to leave the lymph node to induce inflammation.
Both SPNS2 protein and S1P signalling molecule are required for immune cell recruitment to inflammatory organs, which goes towards treating various inflammatory diseases.
“Using pre-clinical models, we have shown that targeting SPNS2 proteins in the body blocks inflammatory responses in disease conditions, such as multiple sclerosis. This work has provided us a possibility to inhibit its transport function with small molecules that will go a long way to treating inflammatory diseases more efficiently and effectively,” said Dr Nguyen.
Colourised electron micrograph image of a macrophage. Credit: NIH
Scientists have created a new treatment for traumatic brain injury (TBI). The new approach leverages macrophages, which can increase or decrease inflammation in response to infection and injury. The team attached “backpacks” containing anti-inflammatory molecules directly to the macrophages. These molecules kept the cells in an anti-inflammatory state when they arrived at the injury site in the brain, enabling them to reduce local inflammation and mitigate the damage caused. The research is reported in PNAS Nexus.
“Every year, millions of people suffer from a TBI, but there is currently no treatment beyond managing symptoms. We have applied our cellular backpack technology – which we previously used to improve macrophages’ inflammatory response to cancerous tumours – to deliver localised anti-inflammatory treatment in the brain, which helps mitigate the cascade of runaway inflammation that causes tissue damage and death in a human-relevant model,” said senior author Samir Mitragotri, PhD, in whose lab the research was performed.
Stopping a runaway inflammation train
There is currently no treatment for the damage caused to brain tissue during a traumatic brain injury (TBI), beyond managing a patient’s symptoms. One of the main drivers of TBI-caused damage is a runaway inflammatory cascade in the brain.
As cells die from the impact, they release a cocktail of pro-inflammatory cytokine molecules that attract immune cells to clean up the damage. But the same cytokine molecules can also disrupt the blood-brain barrier, which causes blood to leak into the brain. Blood accumulation in the brain causes swelling, impaired oxygen delivery, and increased inflammation, and creates a vicious cycle of bleeding and damage that drives even more cell death.
The Mitragotri lab saw an opportunity in this problem.
“It’s generally believed anti-inflammatory therapies can be effective for treating TBI, but so far, none of them have proven effective clinically. Our previous work with macrophages has shown us that we can use our backpack technology to effectively steer their behaviour when they arrive at the injury site. Since these cells are already active players in the body’s natural immune response to a TBI, we had a hunch we could augment that pre-existing biology to reduce the initial damage,” said co-first author Rick Liao, Ph.D., a Postdoctoral Fellow at the Wyss Institute and SEAS.
“Body, heal thyself”…with backpacks
Macrophages are very malleable cells and can “switch” between pro-inflammatory and anti-inflammatory states. While the team’s previous work in cancer had been focused on keeping macrophages in a pro-inflammatory state when they arrive at the inflammation-reducing microenvironment of a tumour, this new project would be trying to do the opposite: keep the macrophages “calm” in the inflammation-riddled setting of a brain injury.
To do so, they used a disc-shaped “backpack” they had previously designed to treat multiple sclerosis that contained layers of two anti-inflammatory molecules: dexamethasone, a steroid, and interleukin-4, a cytokine that encourages macrophages to adopt an anti-inflammatory state. They then incubated these microparticles with both human and pig macrophages in vitro and saw that the backpacks stably stuck to the cells without causing any negative effect. They also observed that application of their backpacks decreased the expression of pro-inflammatory biomarkers and increased the expression of anti-inflammatory biomarkers, retaining the pig macrophages in a healing state.
But to prove that this shift would work in the body, they had to test the backpack-bearing macrophages in vivo. They chose pigs as their model organism because their brains’ structures and responses to injury more closely mimic those of humans than mice.
“Probably our biggest challenge in this project was scaling up production to match what we needed to run the experiments. Our previous studies were done in rodents, which required about two million macrophages and four million backpacks administered per subject. For the porcine study, we needed 100 million macrophages and 200 million backpacks per subject – on the scale of what would be administered in humans – and lots of helping hands,” said co-first author Neha Kapate, PhD, a Postdoctoral Fellow at the Wyss Institute and SEAS.
Once they had generated enough backpack-wearing porcine macrophages, they infused them into the pigs’ bloodstreams four hours after a TBI. Seven days later, they analysed the animals’ brains. Pigs that had received the macrophage treatment showed a high concentration of the cells in the area immediately surrounding the injury site, their lesions were 56% smaller, and there was significantly less haemorrhaging than in untreated animals.
Local immune cells also displayed a lower amount of a pro-inflammatory activation marker called CD80, indicating that the macrophages had accomplished their damage control by reducing inflammation in the brain. Corroborating that data, the levels of two soluble biomarkers for inflammation in the blood and cerebrospinal fluid were lower in treated animals than in untreated animals. The macrophage treatment also did not cause any negative effects.
The team plans to conduct future studies that focus on elucidating exactly how their anti-inflammatory macrophage therapy affects the blood-brain barrier’s integrity to prevent bleeding, which could also hold promise for treating other conditions like hemorrhagic strokes.
“Macrophages’ susceptibility to their local environment has historically prevented scientists from taking full advantage of their immune-modulating capabilities. This impressive study describes a truly novel and potentially powerful macrophage-based therapy for treating the inflammation that is the root cause of so many human afflictions in an effective and non-invasive way that works with biology rather than against it,” said Wyss Founding Director Donald Ingber, MD, PhD.